Sequence Length Distribution
Some high throughput sequencers generate sequence fragments of uniform length, but others can contain reads of wildly varying lengths. Even within uniform length libraries some pipelines will trim sequences to remove poor quality base calls from the end.
This module generates a graph showing the distribution of fragment sizes.
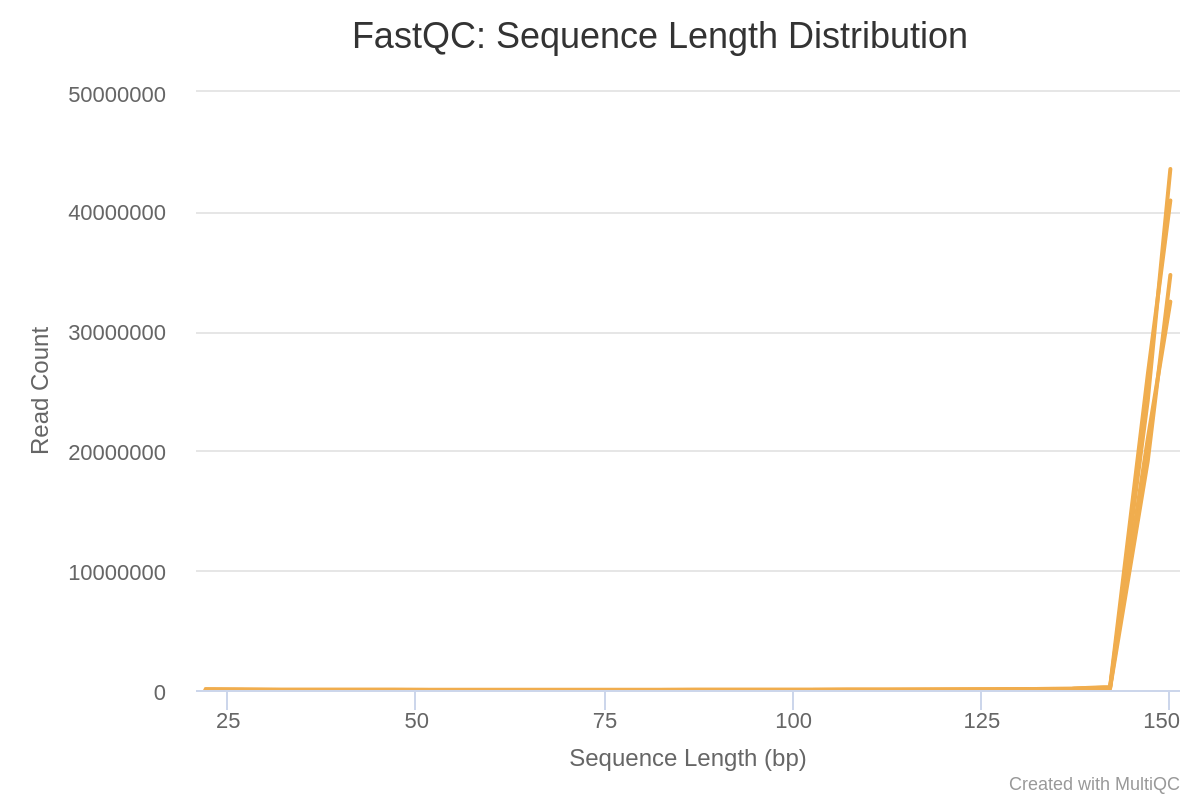
In many cases this will produce a simple graph showing a peak only at one size, but for variable length FastQ files this will show the relative amounts of each different size of sequence fragment.
Warning
This module will raise a warning if all sequences are not the same length.
Failure
This module will raise an error if any of the sequences have zero length.
Common reasons for warnings
Several situations can lead to not unique read lengths (trimming, type of library, sequencing...) so the same observation can be both expected as well as giving rise to a concern regarding the type of library, type of sequencing and data preprocessing applied.
For some sequencing platforms it is entirely normal to have different read lengths so warnings here can be ignored. When sequences have been trimmed to eliminate low quality bases or adapters, this leads to warnings that can also be ignored.
As an example, the following image plot gives expected profiles for trimmed sequences from small RNAseq libraries.
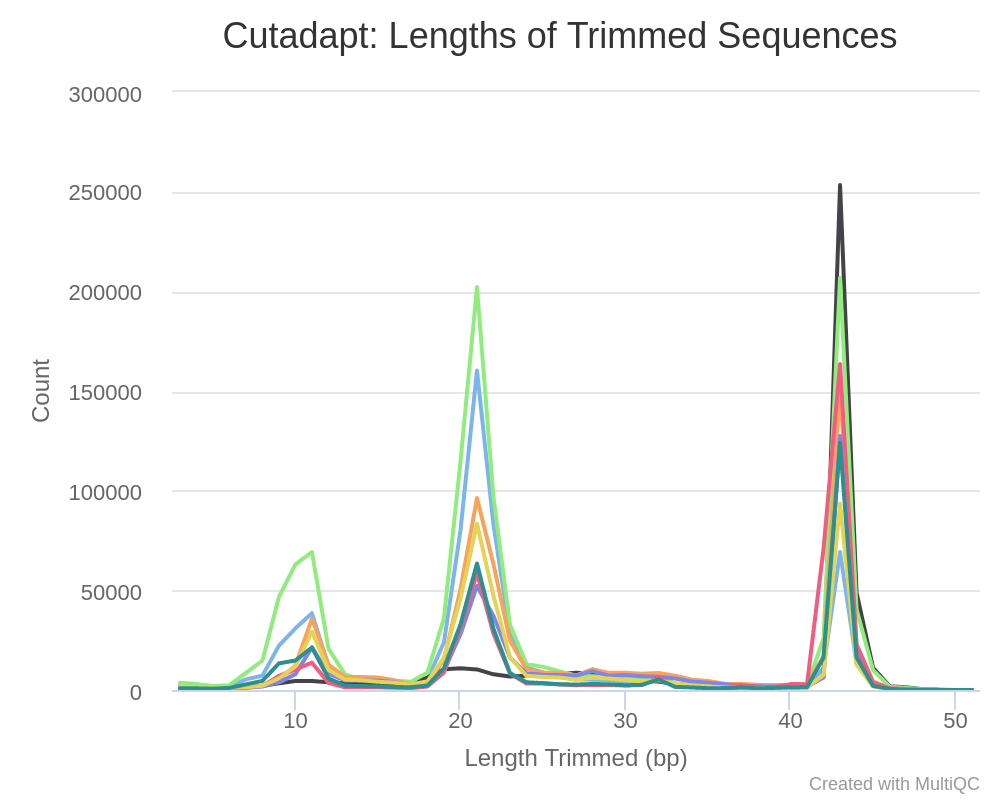
The different peaks correspond to a small RNA biotypes with miRNA corresponding to ~ 22bp peak. But for mRNAseq libraries this kind of profile may be of concern.